ComSin
Description
Download
Search
Statistics
Contact us:
Correspondent author:
Web-service:
Many proteins contain regions without a well-defined ordered structure, called intrinsically disordered regions. Intrinsic disorder has been associated with particular functions including regulation, signaling, and binding of other proteins, nucleic acids and smaller ligands. Many proteins are intrinsically disordered in their native form and fold upon binding. On the other hand, disorder has also been found in the bound state. An exhaustive analysis of intrinsic disorder in protein complexes was done for the first time in our previous paper1. The set of protein structures selected in that work forms the seed of the database which we are presenting now. To gain a clear insight into the abundance of disordered regions in structures of unbound proteins and protein complexes (bound states) as well as disorder-to-order and order-to-disorder transitions upon complex formation, we create an exhaustive database, ComSin, of protein structures in bound ("Complex") and unbound ("Single") states. The usage of this database is not restricted only to the tasks connected with studies of disordered regions in proteins and their complexes. ComSin can be used to analyze any structural differences between proteins in bound and unbound states and to explore changes induced by protein binding.
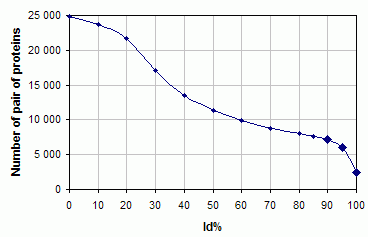 The database consists of pairs of proteins with different levels of identity between the sequences of a chain in unbound
and bound states. We obtained 24910 pairs of complexes and unbound (“single”) proteins. There are 2448, 6051,
and 7129 single-complex
pairs at 100%, 95%, and 90% identity level cutoff, respectively. Our database is designed as following.
The main page contains a description of the information a user may obtain through this database.
On the ComSin search page, there are several filters for selecting a subset of single-complex structure pairs:
"PISA validation", "CBM validation" (for both, please see below), and level of identity between the single and
complex sequences (100%, 95%, and 90%). For example, for 100% identity between bound and unbound states,
one will obtain a list of 2448 pairs of protein structures.
The database consists of pairs of proteins with different levels of identity between the sequences of a chain in unbound
and bound states. We obtained 24910 pairs of complexes and unbound (“single”) proteins. There are 2448, 6051,
and 7129 single-complex
pairs at 100%, 95%, and 90% identity level cutoff, respectively. Our database is designed as following.
The main page contains a description of the information a user may obtain through this database.
On the ComSin search page, there are several filters for selecting a subset of single-complex structure pairs:
"PISA validation", "CBM validation" (for both, please see below), and level of identity between the single and
complex sequences (100%, 95%, and 90%). For example, for 100% identity between bound and unbound states,
one will obtain a list of 2448 pairs of protein structures.
Each line of the obtained list corresponds to the same protein (if 100% identity is selected) or close homologs (if 95% or 90% identity is selected) observed in unbound and bound states. The first column shows the numbering of the pair (unbound protein and its bound homolog) in the current list. The next two columns show the family, according to the Conserved Domains Database2 (CDD), to which both the unbound protein and its bound homolog belong, and its numbering in the current list. By clicking on the CDD family name, a user can see the description of the family at the NCBI web site. In the next column, sequence identity between the unbound protein and its bound homolog is shown. Further, there are three columns related to the structure in the unbound form: the PDB code including the name of the chain, size of the chain, and number of disordered residues in the chain. Similarly, the next three columns correspond to the structure of the bound homolog (PDB code, size of the chain, number of disordered residues). The next two columns correspond to the whole complex. The first column describes type of the complex: it can be a homo- (all chains in the complex are identical) or hetero- (not all chains in the complex are identical) complex. The column C indicates CBM validation of the complex. The final column (P) indicates PISA validation of the complex state.
The CBM3 and PISA4 algorithms were used for validation of the biological relevance of each interaction and oligomeric states. To ensure that interactions observed in the complex (bound state) are biological and not spurious, such as from crystal packing, we use the Conserved Binding Mode (CBM) analysis that confirms interactions by finding several instances of the same domain family pair interacting in the same orientation. PISA validation is based on calculation of the stability of multimeric states inferred from the crystalline state. A link to the new NCBI IBIS (Inferred Biomolecular Interaction Server) database and server is provided which allows the user to look at protein complexes which are homologous to proteins from ComSin database.
The database may also be searched using a PDB code or a CDD family as a query, using the Search link on the main page. Further, a complete list can be downloaded from the main page.
Disordered regions are defined as regions with missing coordinates in X-ray-resolved structures. For each chain, we identify residues with missing coordinates (more precisely, the coordinates of Cα atoms are absent) in the corresponding PDB entry by comparison of the ATOM and SEQRES records (in an X-ray-resolved structure, disordered regions are supposed to be present in SEQRES but absent in ATOM).
By clicking on the number of disordered residues, one is directed to a new page with a comparison of SEQRES and ATOM fields of the corresponding PDB entry. The sequence of the corresponding protein (according to SEQRES and according to ATOM fields) is given in horizontal (short) and vertical (long) format; residues present in SEQRES field but absent in ATOM field (that is, disordered by our definition) are marked in blue. Starting from this page, the user can open the PDB and DSSP files for this protein.
References:
- Fong J, Shoemaker BA, Garbuzynskiy SO, Lobanov MY, Galzitskaya OV, Panchenko AR (2009) Intrinsic disorder in protein interactions: insights from a comprehensive structural analysis. PLoS Comput Biol 5(3):e1000316.
- Marchler-Bauer A, Anderson JB, Derbyshire MK, DeWeese-Scott C, Gonzales NR, Gwadz M, Hao L, He S, Hurwitz DI, Jackson JD, Ke Z, Krylov D, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Thanki N, Yamashita RA, Yin JJ, Zhang D, Bryant SH (2007) CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res 35:D237-40.
- Shoemaker BA, Panchenko AR, Bryant SH (2006) Finding biologically relevant protein domain interactions: conserved binding mode analysis. Protein Sci 15:352-61.
- Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774-97.